


What is Angelman Syndrome?
Angelman Syndrome (AS) is a rare neurogenetic disorder that primarily affects the nervous system. It is a neurodevelopmental condition, meaning it impacts how the brain develops and functions from a very early age. The syndrome is named after Dr. Harry Angelman, who first described the condition in 1965.
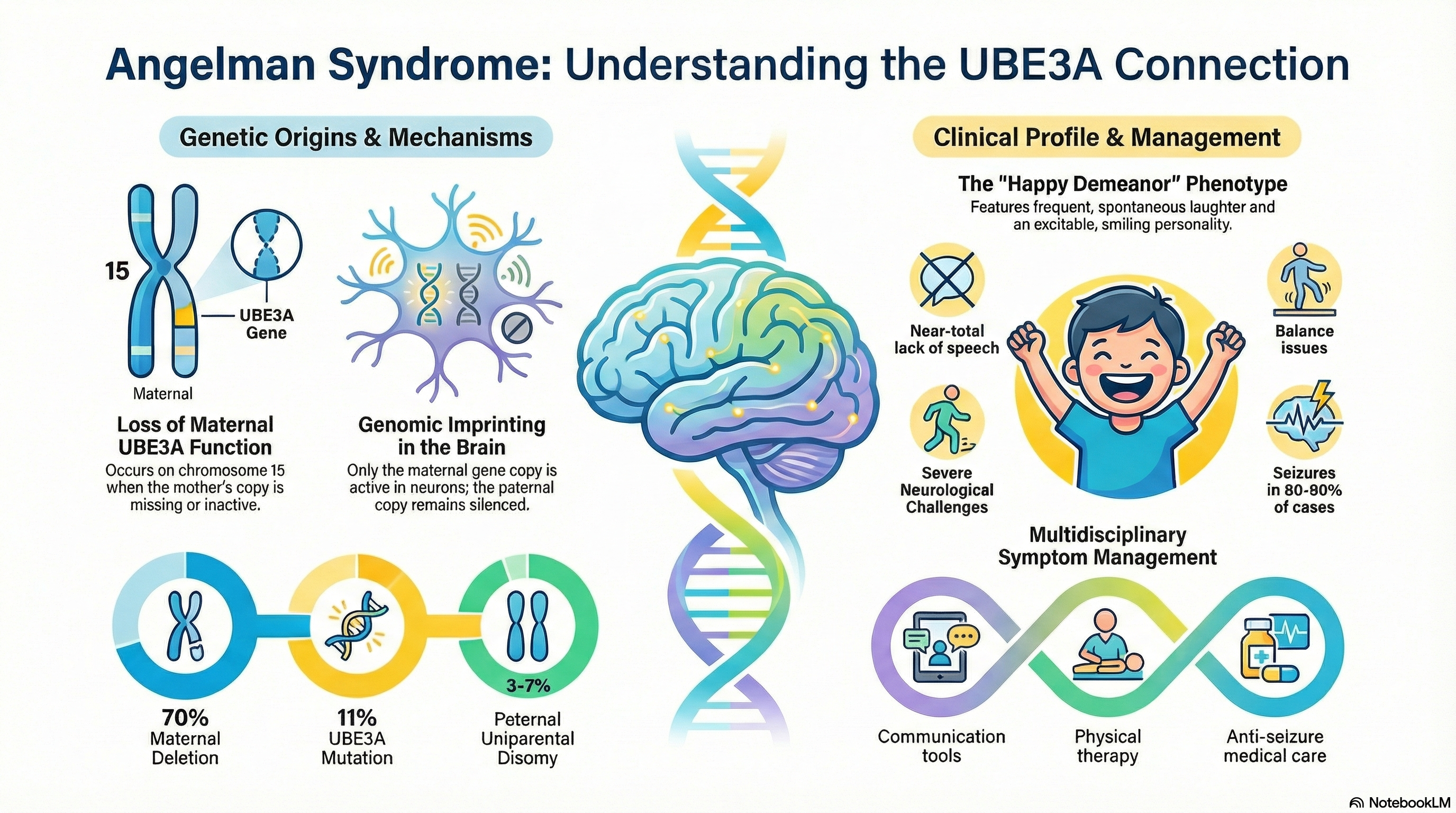
Individuals with Angelman Syndrome typically have a near-normal life expectancy, but they face significant lifelong challenges. The disorder is characterized by a unique combination of severe intellectual disability, a near-total lack of speech, and physical issues with balance and movement. One of the most distinctive features of the syndrome is a "behavioral phenotype" that includes a happy, excitable demeanor with frequent, often spontaneous laughter and smiling.
Causes of Angelman Syndrome
Angelman Syndrome is caused by a loss of function in the maternal copy of the UBE3A gene located on chromosome 15.
This gene is subject to a biological process called genomic imprinting. In most parts of the body, we use copies of genes from both parents. however, in the neurons of the brain, only the copy inherited from the mother is active; the paternal copy is naturally "silenced." If the maternal copy is missing or damaged, the brain has no functioning UBE3A protein to use.
The four main genetic mechanisms include:
-
Maternal Deletion (70%): A small piece of the mother's chromosome 15 is missing. This usually results in the most severe symptoms.
-
UBE3A Mutation (11%): The gene is present but contains a "spelling error" that prevents it from working.
-
Paternal Uniparental Disomy (3-7%): The child inherits two copies of chromosome 15 from the father and none from the mother.
-
Imprinting Defect (3-5%): The maternal gene is present but is incorrectly silenced, just like the paternal copy.
Symptoms of Angelman Syndrome
Symptoms are rarely noticeable at birth. They typically begin to emerge between 6 and 12 months of age as developmental milestones are missed.
Behavioral and Cognitive Symptoms
-
Happy Demeanor: Frequent laughter and smiling, often triggered by minimal stimuli.
-
Speech Impairment: Most individuals do not develop functional speech, though they often understand much more than they can say.
-
Hyperactivity: A very short attention span and a constant state of motion.
-
Hand-Flapping: Repetitive hand movements, especially when excited.
-
Water Fascination: An unusual and persistent love of playing with water or crinkly items like plastic.
Physical and Movement Symptoms
-
Ataxia: Jerky movements and problems with balance and coordination. Walking may be delayed until age 2 or 3 and often appears stiff or wide-based.
-
Seizures: Affecting 80-90% of patients, these typically start between ages 1 and 3.
-
Sleep Issues: A significantly reduced need for sleep and frequent waking throughout the night.
-
Physical Features: These can include a small head size (microcephaly), a wide mouth with widely spaced teeth, and a prominent chin. Some children may have very fair skin and light hair.
Diagnosis of Angelman Syndrome
Diagnosis is often suspected based on the characteristic behavioral and physical signs. However, because symptoms overlap with other conditions like autism or cerebral palsy, genetic testing is required for confirmation.
The Diagnostic Steps Include:
-
DNA Methylation Test: This is the primary screening tool. It identifies whether the maternal UBE3A gene is active. It catches about 80% of cases.
-
Chromosomal Microarray (CMA): This test identifies if a portion of the chromosome is physically missing (deletion).
-
UBE3A Gene Sequencing: If the first tests are normal but symptoms are classic, doctors look for "spelling errors" within the gene itself.
-
EEG (Brain Wave Test): Doctors look for specific, large-scale slow-wave patterns that are very characteristic of Angelman Syndrome, even when the patient is not having a seizure.
Treatment of Angelman Syndrome
There is currently no cure for Angelman Syndrome, so treatment focuses on managing symptoms and supporting development through a multidisciplinary team.
Therapeutic Support
-
Communication Training: Using non-verbal tools such as iPads with communication apps, picture exchange systems (PECS), and sign language.
-
Physical and Occupational Therapy: Focusing on improving gait, balance, and fine motor skills (like feeding and dressing).
-
Behavioral Therapy: Helping to manage hyperactivity and improve the attention span.
Medical Management
-
Seizure Control: Anti-seizure medications are a cornerstone of treatment.
-
Sleep Aids: Melatonin or other prescribed sedatives are frequently used to help regulate sleep cycles.
-
GI Support: Management of common issues like acid reflux (GERD) and chronic constipation.
-
Scoliosis Monitoring: Regular check-ups for spine curvature, which may require bracing or surgery in adolescence.
Prevention of Angelman Syndrome
Most cases of Angelman Syndrome are not inherited from a parent; they occur as a random genetic "accident" during the formation of reproductive cells or very early in the development of the embryo. In these cases, the risk of a second child having the condition is extremely low (less than 1%).
However, certain forms of the disorder—specifically UBE3A mutations and imprinting defects—can be passed down through families. If a mother carries one of these genetic changes, there may be a 50% risk of recurrence in future children.
Prevention and Planning involve:
-
Genetic Counseling: Families with one affected child should meet with a geneticist to determine the specific genetic mechanism involved and understand the risks for future pregnancies.
-
Prenatal Testing: For families at high risk, prenatal diagnostic tests are available to check for the UBE3A status of the fetus.
-
Early Surveillance: While the syndrome itself cannot be prevented if the genetic change is present, "preventing" secondary complications (like severe dental issues or advanced scoliosis) is possible through early and consistent medical intervention.
Whenever possible, families are encouraged to join support organizations to stay informed about emerging gene-based therapies currently in clinical trials that aim to reactivate the silent paternal gene.


